
概要
ミトコンドリア病は、細胞内のエネルギー代謝に欠陥が生じることによって引き起こされる、極めて多様性のある遺伝性疾患群である。
ミトコンドリアはすべてのヒト細胞に存在する重要な細胞小器官であり、その機能不全は体のあらゆる臓器に影響を及ぼす可能性がある。特に、脳・心筋・骨格筋など、高いエネルギー需要のある組織が脆弱であり、神経学的症状が顕著に現れることが多い。
小児期に発症する重症例から、成人期後期に発症する比較的軽症な例まで、あらゆる年齢で、あらゆる症状を呈することが特徴である。生涯リスクは、およそ1,500人に1人である。
病因
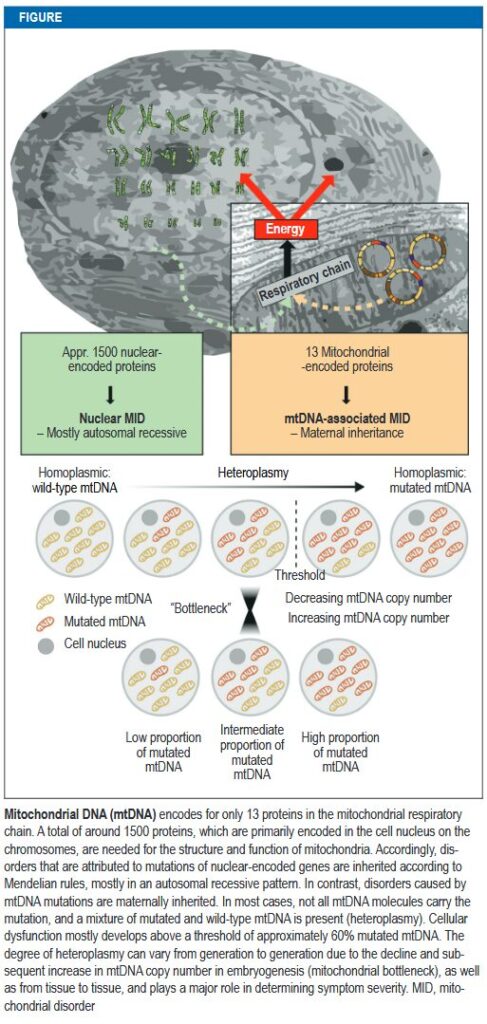
ミトコンドリアの構造と機能は、約1,500のタンパク質の相互作用によって決定され、エネルギー代謝の病原性欠陥は、これまでに400以上の関連遺伝子に起因することが確認されている。ミトコンドリアは核DNA(nDNA)とミトコンドリアDNA(mtDNA)の二重の遺伝的制御を受けている。
*Dtsch Arztebl Int. 2021;118(44):741-748.
- ミトコンドリアDNA (mtDNA) の関与
-
- 母系遺伝:mtDNAは母親からのみ子孫に遺伝する。精子に含まれる少数のミトコンドリアは受精時に排除されるため、父親のmtDNAは遺伝に寄与しない。
- ホモプラスミーとヘテロプラスミー:個々のヒト細胞は通常遺伝的に同一な数千のmtDNAコピーを持っている(ホモプラスミー)。mtDNAに変異を持つミトコンドリア病の患者は、正常と変異の両方のmtDNAを個々の細胞に保持している(ヘテロプラスミー)。
- 閾値効果:ヘテロプラスミーの場合、変異mtDNAの割合が一定の閾値を超えると、臨床症状が発現すると考えられている。この閾値は組織によって異なり、変異の割合が世代間で大きく変動するため、同じ家族内でも症状の重症度にばらつきが生じる。
- 原因遺伝子:mtDNAは、呼吸鎖の13の構造タンパク質、22のtRNA、2つのrRNAをコードしている。これらの37遺伝子すべてに病原性変異が記述されている。mtDNA変異による成人発症型疾患は、約3分の2を占めるとの報告がある。
- 核DNA (nDNA) の関与
-
- メンデル遺伝:ミトコンドリアのタンパク質の大部分(約1,500個)は核DNAによってコードされており、これらの遺伝子変異はメンデルの法則に従って遺伝する。ほとんどは常染色体潜性遺伝、一部常染色体顕性遺伝やX染色体性遺伝もある。
- 原因遺伝子:核DNAには、酸化リン酸化システム (OXPHOS) のサブユニット、ミトコンドリアDNAの複製、転写、維持に必要なタンパク質などをコードする350以上の遺伝子が存在する。これらの欠陥は、mtDNAの枯渇や多発性mtDNA欠失を引き起こすことがある。
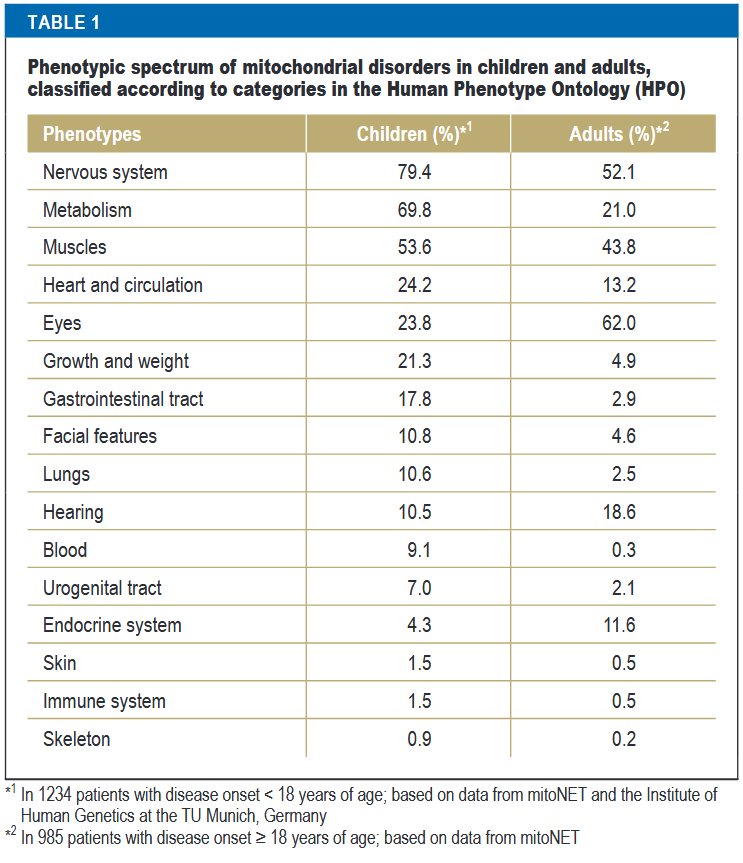
臨床症候
ミトコンドリアはすべての細胞に不可欠な細胞小器官であるため、ミトコンドリア病はあらゆる臓器に影響を及ぼす可能性があり、その症状は多岐にわたる。神経系の関与が顕著なことが多いが、神経学的症状がない、またはそれ以上に全身症状が優位な場合もある。
*Dtsch Arztebl Int. 2021;118(44):741-748.
神経症状(急性・亜急性)
- 脳卒中様発作(Stroke-like episodes)
-
ミトコンドリア病、特にMELASの定義的特徴である。多くは40歳未満で発症するが、高齢発症も報告されている。頭痛・嘔気・視覚異常といった前駆症状に続き、けいれん・意識障害・局所神経脱落症状が出現する。
代表的な遺伝子変異:m.3243A>G(MELAS症例の最大80%を占める)・POLG変異
画像所見:MRIでは特定の血管支配領域に一致しない大脳皮質および皮質下白質の病変が特徴的。
鑑別診断:非定型的な若年性脳梗塞、感染性・自己免疫性脳炎、PRES
- てんかん
-
脳卒中様発作に密接に関連し、しばしばその誘因ともなる。全般発作と焦点発作の両方が見られる。特に脳卒中様発作に伴うけいれんは治療抵抗性となることが多く、POLG遺伝子関連てんかんでは、一般的な抗発作薬のみならず麻酔薬に対しても薬剤抵抗性を示すことがあり、臨床管理上の大きな課題となる。
代表的な遺伝子変異:m.3243A>G、POLG
- 脳幹機能の代謝性代償不全
-
主に小児期のミトコンドリア病で最も一般的な病型であるLeigh症候群は、軽症例では成人期まで生存することがある。これらの患者は、感染症などを契機として、眼球運動障害・嚥下障害・構音障害・中枢性低換気といった亜急性の脳幹機能不全を呈することがある。
代表的な遺伝子変異:MT-ATP6、SURF1など多数
- 亜急性の視神経症
-
レーベル遺伝性視神経症(LHON)の典型的な症状である。通常、片眼の無痛性かつ進行性の中心視力障害として発症し、数週間から数ヶ月以内にもう一方の眼にも同様の症状が現れる。
鑑別診断: 視神経炎(特に非典型的な経過をたどる場合)
神経症状(慢性進行性)
- 慢性進行性外眼筋麻痺 (Chronic progressive external ophthalmoplegia:CPEO)
-
成人発症のミトコンドリア病で最も一般的な臨床像の一つである。緩徐に進行する眼瞼下垂と外眼筋の麻痺が特徴である。進行が非常に遅いため、患者が複視を訴えることは比較的少ない(30%未満)とされている。
鑑別診断:重症筋無力症、眼咽頭型筋ジストロフィー、先天性ミオパチー
- ミオパチー
-
易疲労性・運動不耐性・運動誘発性の筋痛が主症状である。クレアチンキナーゼ(CK)の上昇は軽度であることが多いが、時に1000 IU/Lを超えることもある。
- 感音難聴
-
若年発症で、母系遺伝の家族歴を伴う場合、ミトコンドリア病を強く示唆する。特定のmtDNA変異(例:m.1555A>G)では、アミノグリコシド系抗生物質への曝露が難聴の引き金となることがある。
- 視神経症
-
OPA1遺伝子変異による常染色体優性視神経萎縮症が代表的である。小児期発症で緩徐に進行する両側性の視神経萎縮を特徴とする。患者の約20%は、難聴、CPEO、運動失調、パーキンソニズムなど、視神経以外の症状を合併する。
- その他
-
小脳性・感覚性の運動失調、末梢神経障害、片頭痛、認知機能障害、痙性、錐体外路症状(振戦・パーキンソニズム・ジストニア・舞踏運動)
その他
心臓:肥大型心筋症、不整脈 (伝導障害)
内分泌:糖尿病、副甲状腺機能低下症、低身長
消化器:偽性腸閉塞、嚥下障害、吸収不良、悪心・嘔吐
肝臓:肝機能障害
腎臓:腎尿細管機能障害(Fanconi症候群)
血液:汎血球減少症、鉄芽球性貧血
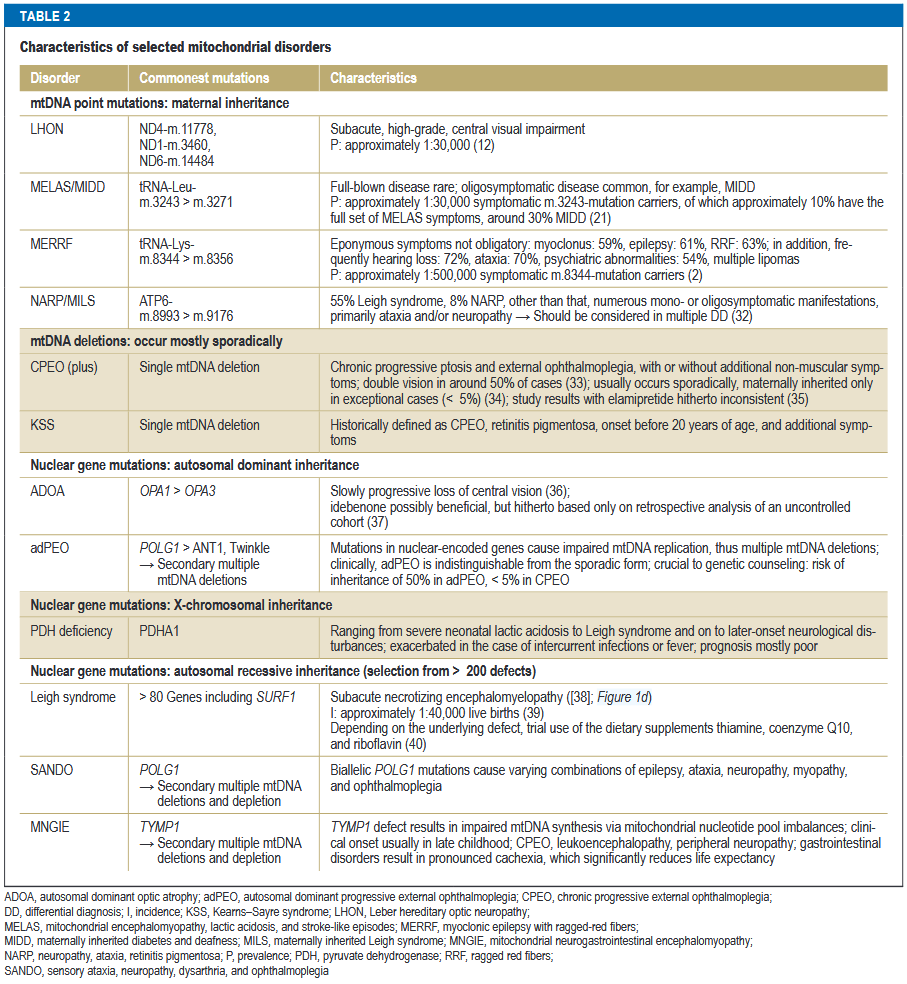
主な病型
多くの患者は1つのカテゴリーに当てはめることができず、症候群のオーバーラップを呈する。
*Dtsch Arztebl Int. 2021;118(44):741-748.
MELAS:mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes
脳卒中様エピソード:頭痛・嘔気嘔吐・脳症・焦点性てんかん発作・精神症状に引き続いて神経脱落症候が出現する。通常10~20代で発症。
原因遺伝子:m.3243A>G変異が症例の最大80%を占める。
MRI:脳卒中様病変が通常血管支配領域に対応せず、皮質や皮質下白質に現れる。MRSでは病変部・脳室内・病変部以外のいずれでも乳酸ピークがみられる。病変は数日~数週あるいは数か月かけて拡大し、その後は消失することもあるが、皮質の層状壊死・グリオーシス・萎縮をきたすことが多い。
糖尿病や難聴も高頻度にみられる。
リー症候群(Leigh syndrome)
小児ミトコンドリア病で最も一般的な病型。代謝ストレス時に亜急性脳幹機能障害を発症。乳児期に精神運動発達遅滞・筋緊張低下・運動失調・呼吸不全・不随意運動を伴う進行性神経変性疾患。
原因遺伝子:m.8993T>C MT-ATP6変異やSURF1遺伝子(AR)の変異など、80以上の遺伝子欠陥が関連する。
画像所見:脳幹機能障害では、髄質の非対称病変や基底核の対称性T2高信号が見られることがある。
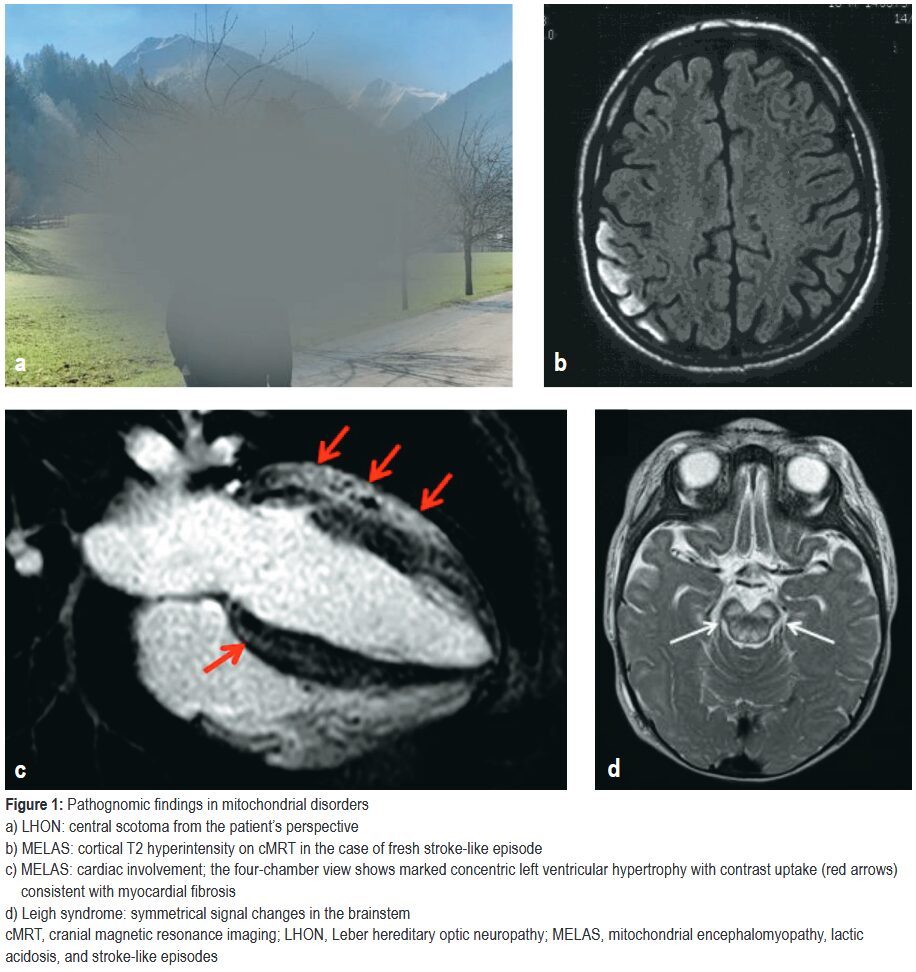
レーベル遺伝性視神経症(LHON:Leber’s hereditary optic neuropathy)
亜急性の無痛性進行性中心視力障害。通常片眼から始まり、数週から数ヶ月で両眼が影響を受ける。若年男性に好発する。喫煙と過剰なアルコール摂取が視力低下の誘発因子となる。
原因遺伝子:mtDNAのMT-ND1, MT-ND4, MT-ND6遺伝子の3つの主要な病原性変異 (m.3460G→A, m.11778G→A, m.14484T→C) が90%以上の症例を占める。
慢性進行性外眼筋麻痺(CPEO:chronic progressive external ophthalmoplegia)
成人期ミトコンドリア病で最も一般的な臨床像の一つ。徐々に進行する両側性の眼瞼下垂と眼球運動制限が典型的で、約50%の患者で複視を伴う。
原因遺伝子:mtDNAの単一大規模欠失が最も多いが、核DNA変異による多発性mtDNA欠失の場合もある。
Kearns-Sayre syndrome(KSS):CPEO・色素性網膜症・心伝導障害(ペースメーカーを要することが多い)を三徴とする重篤な病型。
MERRF:myoclonic epilepsy associated with ragged red fiber
ミオクローヌスを伴うてんかん・運動失調・ミオパチーが特徴。難聴・認知機能低下の頻度も高い。体幹(特に肩甲帯)や頚部に多発性脂肪腫がみられることがある。
原因遺伝子:MT-TK遺伝子のm.8344A>G変異が症例の80%を占める。
検査
乳酸(Lactate)
解糖系から生じたピルビン酸がミトコンドリア内で十分に利用されない場合、乳酸の産生が亢進することで乳酸値が上昇する。
血中乳酸値の上昇(>2mmol/L)はミトコンドリアの機能不全を示唆する。氷上管理をしない、止血帯使用をするなどのサンプリングの問題で偽陽性になることが多い。他の代謝異常や組織虚血などによっても上昇する。静脈採血で高値であった場合は、動脈採血で再検が必要。反復採血により恒常的な高乳酸血症を確認する。感度34-62%・特異度83-100%との報告あり。食後の乳酸値評価によって感度は上昇する。
神経症状を呈している例では、髄液中の乳酸値の上昇はミトコンドリア病の診断マーカーとして有用。しかし、てんかん重積などの他病態でも上昇することがあることに留意する。
ピルビン酸(Pyruvate)
血中ピルビン酸:感度34.6-88.2%・特異度81.2-87.2%
髄液ピルビン酸:感度88.2%・特異度100%
との報告あり。
乳酸/ピルビン酸比(L/P ratio)
乳酸値が高値である場合に評価する。
ピルビン酸の代謝異常などでも乳酸・ピルビン酸の上昇がみられるが、その場合はL/P比は正常となる。一方でミトコンドリア病では乳酸がピルビン酸に比べて相対的に高くなるため、L/P比が高値となることから、これらの病態を区別するために有用。
正常では10-15未満、15-20以上で上昇と判断。
髄液L/P比の上昇:感度76.5%・特異度90.6%との報告あり。
画像検査
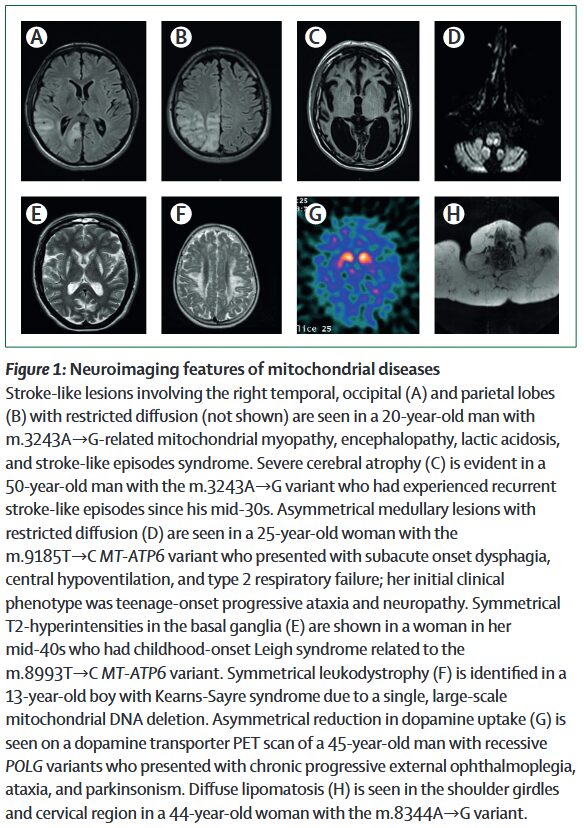
*Lancet Neurol. 2021;20(7):573-584.
ゲノム解析
mtDNA:成人発症のミトコンドリア病の約3分の2はmtDNA変異が原因とされる。全mtDNAゲノムの網羅的解析が可能。血液・尿沈渣・口腔粘膜スワブなどの非侵襲的検体から微量な変異(低ヘテロプラスミー率)を正確に定量することができる。組織特異的な変異である場合は、血液のみならず傷害組織の生検検体(骨格筋や肝臓など)の解析が必要となる。
核DNA:ミトコンドリア病の原因となる核遺伝子は350以上同定されている。網羅的に遺伝子を解析する全エクソーム解析・全ゲノム解析を用いる。
筋生検
かつては診断の中心であったが、ゲノム解析が可能となったため、現在は遺伝子診断で不確定な例に対して用いる最終手段としての立ち位置。筋組織特異的なmtDNA異常の検出やその他の鑑別疾患の評価を中心に行う。
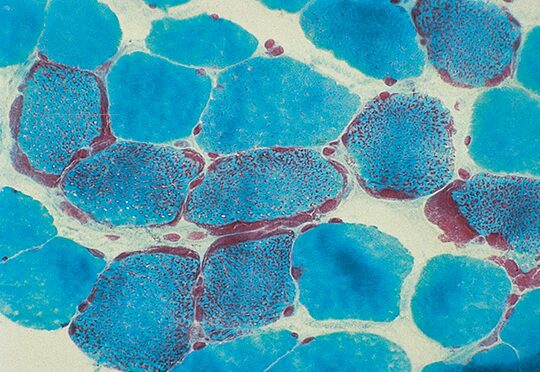
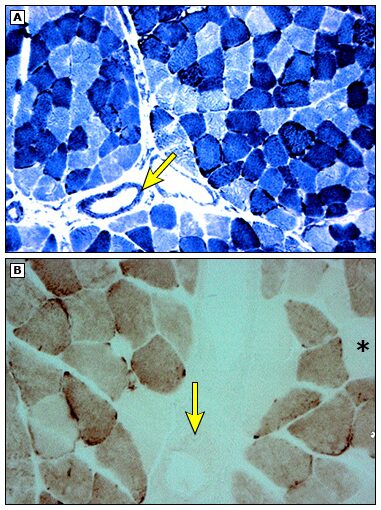
ミトコンドリア病の特徴としては、modified Gomori Trichrome染色で増生した異常ミトコンドリアが赤色ぼろ線維(RRF:ragged red fiber)として確認できる。ミトコンドリアを特異的に染色するコハク酸脱水素酵素(SDH:succinate dehydrogenase)の活性染色で濃染する。チトクロームC酸化酵素(COX:cytochrome C oxidase)染色で染色性を欠く線維やSDHの活性染色で動脈壁の濃染(SSV:strongly SDH-reactive blood vessels)も本症を疑う根拠となる。
- mGT染色
-

- A:SDH染色(矢印はSSVの所見)
B:COX染色(*は染色陰性の筋線維、矢印はSSVのCOX陰性所見) -
診断
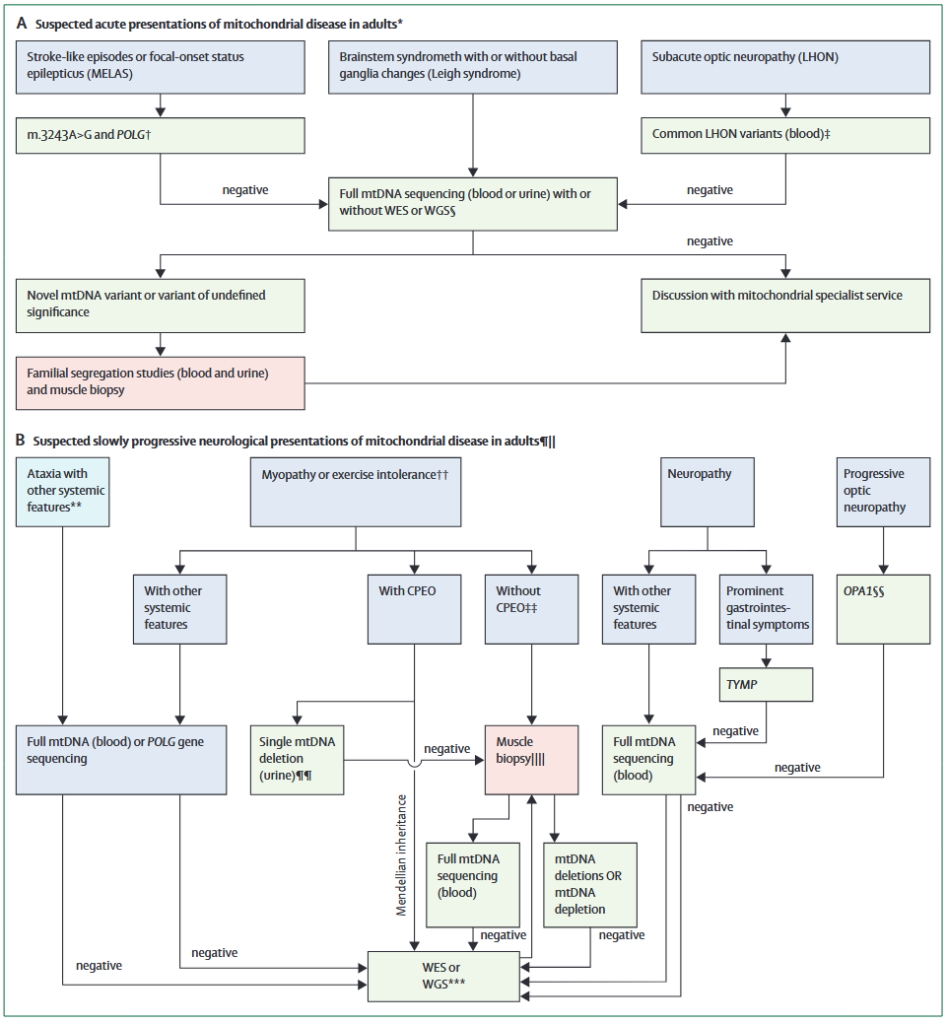
*Lancet Neurol. 2021;20(7):573-584.
厚生労働省の診断基準
- 1.主症状
-
①進行性の筋力低下、横紋筋融解、または外眼筋麻痺を認める。
②知的退行、記銘力障害、けいれん、精神症状、一過性麻痺、半盲、皮質盲、ミオクローヌス、ジストニア、小脳失調などの中枢神経症状のうち、1つ以上を認める。または手足のしびれなどの末梢神経障害を認める。
③心伝導障害・心筋症などの心症状、肺高血圧症などの呼吸器症状、糸球体硬化症・腎尿細管機能異常などの腎症状、強度の貧血などの血液症状、肝障害・黄疸・凝固能低下などの肝症状、体重増加不良・繰り返す嘔吐/下痢/便秘などの消化器症状を認める。
④低身長・甲状腺機能低下症などの内分泌症状や糖尿病を認める。
⑤強度視力低下・網膜色素変性などの眼症状、感音難聴などの耳症状を認める。
⑥新生児期または乳児期に、発育異常・発達遅延を認める。
- 2.検査・画像所見
-
①ミトコンドリアDNAの質的・量的異常、またはミトコンドリア関連分子をコードする核遺伝子変異を認める。
②骨格筋生検・培養細胞・症状のある臓器の細胞組織で、ミトコンドリア病に特徴的な病理所見や電子顕微鏡所見を認める。
③ミトコンドリア関連酵素の活性低下、またはコエンザイムQ10などの中間代謝物の欠乏を認める。または、ミトコンドリアDNAの発現異常を認める。
④安静臥床時の血液または髄液の乳酸値が繰り返して高い、またはMRSで病変部に明らかな乳酸ピークを認める。
⑤脳CT/MRIにて、大脳基底核・脳幹に両側対称性の病変等を認める。
⑥眼底検査にて、急性期においては蛍光漏出を伴わない視神経乳頭の発赤・腫脹、視神経乳頭近傍毛細血管蛇行、網膜神経線維腫大、視神経乳頭近傍の出血のうち1つ以上の所見を認めるか、慢性期(視力低下の発症から通常6か月以降)における視神経萎縮所見を両眼に認める。
⑦腹部エコー、CT、MRIあるいは肝組織所見にて、脂肪肝あるいは肝硬変の所見を認める。
- Definite
-
(1)①~⑤のうち1項目以上あり、かつ(2)①を満たす。
(1)①~⑤のうち1項目以上あり、かつ(2)②~③と④~⑦のそれぞれで1項目以上を満たす。
- Probable
-
(1)⑥があり、かつ(2)①を満たす。
(1)①~⑥のうち1項目以上あり、かつ(2)②~③のうち1項目以上を満たす。
(1)①~⑥のうち1項目以上あり、かつ(2)④~⑦のうち2項目以上を満たす。
- Possible
-
(1)①~⑥のうち1項目以上あり、かつ(2)④~⑥のうち1項目以上を満たす。
引用・参考文献
Dtsch Arztebl Int. 2021;118(44):741-748.
Lancet Neurol. 2021;20(7):573-584.
Genet Med. 2015;17(9):689-701.
Ann Transl Med. 2018;6(24):475.

